Chapter 4 Selection scheme diagnostic experiments
experiment_slug <- "2023-05-10-diagnostics"
working_directory <- paste0(
"experiments/",
experiment_slug,
"/analysis/"
)
if (exists("bookdown_wd_prefix")) {
working_directory <- paste0(
bookdown_wd_prefix,
working_directory
)
}4.1 Dependencies
library(tidyverse)## ── Attaching packages ──────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse 1.3.2 ──
## ✔ ggplot2 3.3.6 ✔ purrr 1.0.1
## ✔ tibble 3.1.8 ✔ dplyr 1.1.0
## ✔ tidyr 1.3.0 ✔ stringr 1.5.0
## ✔ readr 2.1.3 ✔ forcats 0.5.2
## ── Conflicts ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()library(ggplot2)
library(cowplot)
library(RColorBrewer)
library(khroma)
library(rstatix)##
## Attaching package: 'rstatix'
##
## The following object is masked from 'package:stats':
##
## filterlibrary(knitr)
source("https://gist.githubusercontent.com/benmarwick/2a1bb0133ff568cbe28d/raw/fb53bd97121f7f9ce947837ef1a4c65a73bffb3f/geom_flat_violin.R")print(version)## _
## platform aarch64-apple-darwin20
## arch aarch64
## os darwin20
## system aarch64, darwin20
## status
## major 4
## minor 2.1
## year 2022
## month 06
## day 23
## svn rev 82513
## language R
## version.string R version 4.2.1 (2022-06-23)
## nickname Funny-Looking Kid4.2 Setup
# Configure our default graphing theme
theme_set(theme_cowplot())
# Create a directory to store plots
plot_directory <- paste0(working_directory, "plots/")
dir.create(plot_directory, showWarnings=FALSE)4.2.1 Load experiment summary data
summary_data_loc <- paste0(working_directory, "data/aggregate.csv")
summary_data <- read_csv(summary_data_loc)## Rows: 520 Columns: 49
## ── Column specification ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (5): DIAGNOSTIC, EVAL_FIT_EST_MODE, EVAL_MODE, SELECTION, STOP_MODE
## dbl (44): ACCURACY, CREDIT, DIAGNOSTIC_DIMENSIONALITY, GENE_LOWER_BND, GENE_...
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.summary_data <- summary_data %>%
mutate(
eval_mode_row = case_when(
EVAL_MODE == "full" & TEST_DOWNSAMPLE_RATE == "1" ~ "down-sample",
EVAL_MODE == "full" & NUM_COHORTS == "1" ~ "cohort",
.default = EVAL_MODE
),
evals_per_gen = case_when(
EVAL_MODE == "cohort-full-compete" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "cohort" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "down-sample" ~ TEST_DOWNSAMPLE_RATE,
EVAL_MODE == "full" ~ 1.0
),
EVAL_FIT_EST_MODE = case_when(
EVAL_FIT_EST_MODE == "ancestor-opt" ~ "ancestor",
EVAL_FIT_EST_MODE == "relative-opt" ~ "relative",
.default = EVAL_FIT_EST_MODE
),
.keep = "all"
) %>%
mutate(
evals_per_gen = as.factor(evals_per_gen),
eval_mode_row = as.factor(eval_mode_row),
DIAGNOSTIC = as.factor(DIAGNOSTIC),
SELECTION = as.factor(SELECTION),
EVAL_MODE = as.factor(EVAL_MODE),
NUM_COHORTS = as.factor(NUM_COHORTS),
TEST_DOWNSAMPLE_RATE = as.factor(TEST_DOWNSAMPLE_RATE),
EVAL_FIT_EST_MODE = factor(
EVAL_FIT_EST_MODE,
levels = c(
"none",
"ancestor",
"relative"
),
labels = c(
"None",
"Ancestor",
"Relative"
)
)
)
# Split summary data on diagnostic
con_obj_summary_data <- filter(
summary_data,
DIAGNOSTIC == "contradictory-objectives"
)
explore_summary_data <- filter(
summary_data,
DIAGNOSTIC == "multipath-exploration"
)4.2.2 Load experiment time series data
ts_data_loc <- paste0(working_directory, "data/time_series.csv")
ts_data <- read_csv(ts_data_loc)## Rows: 104000 Columns: 19
## ── Column specification ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (4): DIAGNOSTIC, EVAL_FIT_EST_MODE, EVAL_MODE, SELECTION
## dbl (15): NUM_COHORTS, SEED, TEST_DOWNSAMPLE_RATE, entropy_selected_ids, eva...
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.ts_data <- ts_data %>%
mutate(
eval_mode_row = case_when(
EVAL_MODE == "full" & TEST_DOWNSAMPLE_RATE == "1" ~ "down-sample",
EVAL_MODE == "full" & NUM_COHORTS == "1" ~ "cohort",
.default = EVAL_MODE
),
evals_per_gen = case_when(
EVAL_MODE == "cohort-full-compete" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "cohort" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "down-sample" ~ TEST_DOWNSAMPLE_RATE,
EVAL_MODE == "full" ~ 1.0
),
EVAL_FIT_EST_MODE = case_when(
EVAL_FIT_EST_MODE == "ancestor-opt" ~ "ancestor",
EVAL_FIT_EST_MODE == "relative-opt" ~ "relative",
.default = EVAL_FIT_EST_MODE
),
.keep = "all"
) %>%
mutate(
evals_per_gen = as.factor(evals_per_gen),
DIAGNOSTIC = as.factor(DIAGNOSTIC),
SELECTION = as.factor(SELECTION),
EVAL_MODE = as.factor(EVAL_MODE),
NUM_COHORTS = as.factor(NUM_COHORTS),
TEST_DOWNSAMPLE_RATE = as.factor(TEST_DOWNSAMPLE_RATE),
EVAL_FIT_EST_MODE = factor(
EVAL_FIT_EST_MODE,
levels = c(
"none",
"ancestor",
"relative"
),
labels = c(
"None",
"Ancestor",
"Relative"
)
)
)
con_obj_ts_data <- ts_data %>%
filter(DIAGNOSTIC == "contradictory-objectives")
explore_ts_data <- ts_data %>%
filter(DIAGNOSTIC == "multipath-exploration")4.2.3 Load estimate source distributions
est_source_data <- read_csv(
paste0(working_directory, "data/phylo-est-info.csv")
)## Rows: 520 Columns: 38
## ── Column specification ─────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────────
## Delimiter: ","
## chr (6): OUTPUT_DIR, DIAGNOSTIC, STOP_MODE, SELECTION, EVAL_MODE, EVAL_FIT_...
## dbl (32): SNAPSHOT_INTERVAL, OUTPUT_SUMMARY_DATA_INTERVAL, MUTATE_STD, TARGE...
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.est_source_data <- est_source_data %>%
mutate(
eval_mode_row = case_when(
EVAL_MODE == "full" & TEST_DOWNSAMPLE_RATE == "1" ~ "down-sample",
EVAL_MODE == "full" & NUM_COHORTS == "1" ~ "cohort",
.default = EVAL_MODE
),
evals_per_gen = case_when(
EVAL_MODE == "cohort-full-compete" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "cohort" ~ 1.0 / NUM_COHORTS,
EVAL_MODE == "down-sample" ~ TEST_DOWNSAMPLE_RATE,
EVAL_MODE == "full" ~ 1.0
),
EVAL_FIT_EST_MODE = case_when(
EVAL_FIT_EST_MODE == "ancestor-opt" ~ "ancestor",
EVAL_FIT_EST_MODE == "relative-opt" ~ "relative",
.default = EVAL_FIT_EST_MODE
),
.keep = "all"
) %>%
mutate(
evals_per_gen = as.factor(evals_per_gen),
eval_mode_row = as.factor(eval_mode_row),
DIAGNOSTIC = as.factor(DIAGNOSTIC),
SELECTION = as.factor(SELECTION),
EVAL_MODE = as.factor(EVAL_MODE),
NUM_COHORTS = as.factor(NUM_COHORTS),
TEST_DOWNSAMPLE_RATE = as.factor(TEST_DOWNSAMPLE_RATE),
EVAL_FIT_EST_MODE = factor(
EVAL_FIT_EST_MODE,
levels = c(
"none",
"ancestor",
"relative"
),
labels = c(
"None",
"Ancestor",
"Relative"
)
)
) %>%
mutate(
prop_self_lookups = case_when(
(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") ~
self_count / (other_count + ancestor_count + descendant_count + self_count + outside_count),
.default = 0
),
prop_other_lookups = case_when(
(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") ~
other_count / (other_count + ancestor_count + descendant_count + self_count + outside_count),
.default = 0
),
prop_ancestor_lookups = case_when(
(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") ~
ancestor_count / (other_count + ancestor_count + descendant_count + self_count + outside_count),
.default = 0
),
prop_descendant_lookups = case_when(
(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") ~
descendant_count / (other_count + ancestor_count + descendant_count + self_count + outside_count),
.default = 0
),
prop_outside_lookups = case_when(
(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") ~
outside_count / (other_count + ancestor_count + descendant_count + self_count + outside_count),
.default = 0
)
)4.3 Contradictory objectives diagnostic
4.3.1 Population-wide satisfactory trait coverage (final)
Satifactory trait coverage after 50,000 generations:
contradictory_obj_final_plt <- ggplot(
con_obj_summary_data,
aes(
x = EVAL_FIT_EST_MODE,
y = pop_optimal_trait_coverage,
fill = EVAL_FIT_EST_MODE
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
adjust=1.5
) +
geom_point(
mapping=aes(color=EVAL_FIT_EST_MODE),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
# limits = c(-0.5, 100)
) +
scale_fill_bright() +
scale_color_bright() +
facet_grid(
eval_mode_row~evals_per_gen,
# nrow=2,
labeller=label_both
) +
theme(
legend.position = "none",
axis.text.x = element_text(
angle = 30,
hjust = 1
),
panel.border = element_rect(color="gray", size=2)
)
ggsave(
filename = paste0(plot_directory, "contra-obj-final.pdf"),
plot = contradictory_obj_final_plt + labs(title="Contradictory objectives"),
width = 15,
height = 10
)contradictory_obj_final_plt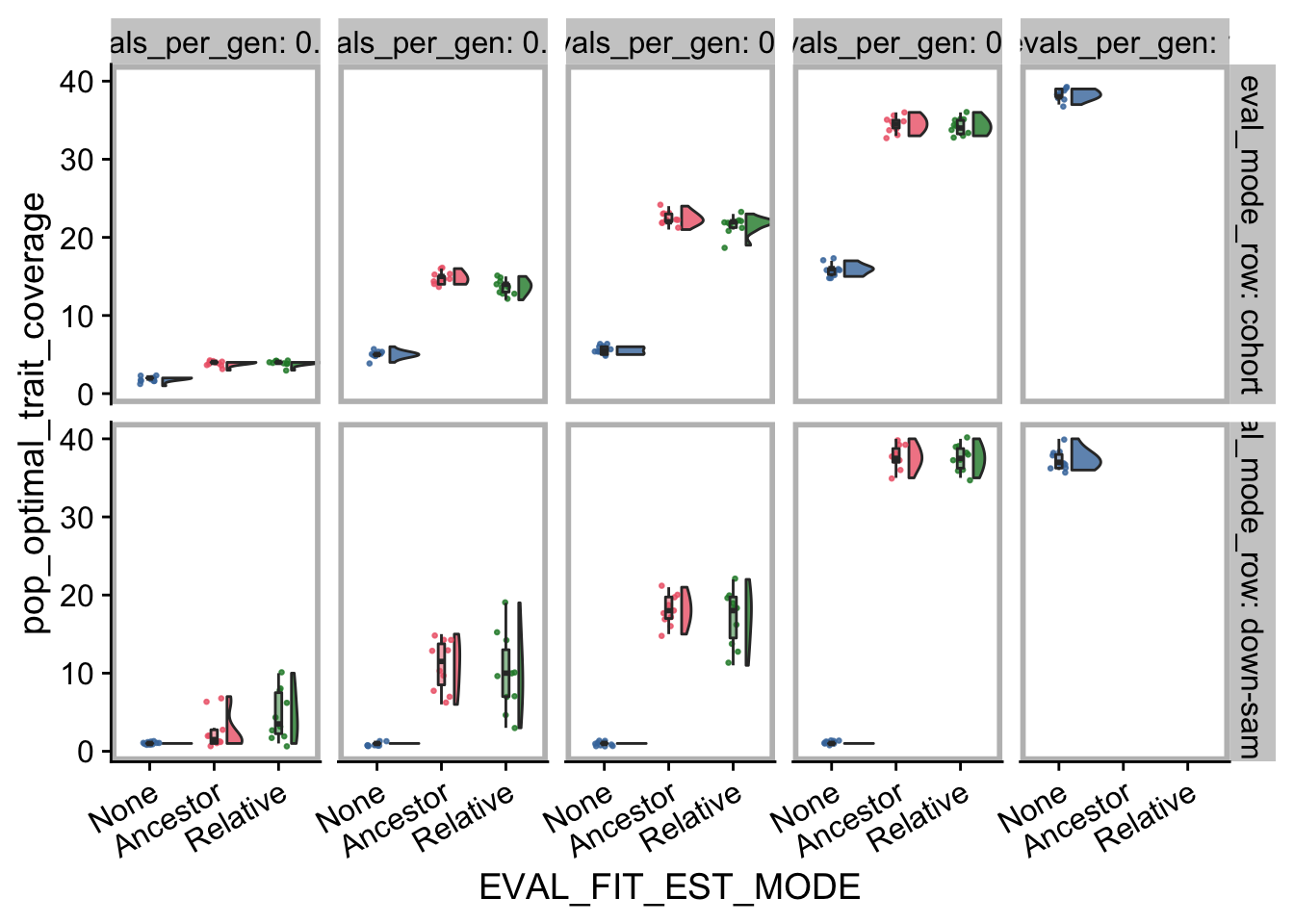
4.3.1.1 Statistical analysis
First, we create a table of summary values for satisfactory trait coverage in the final generation.
con_obj_summary_data %>%
filter(EVAL_MODE != "full") %>%
group_by(EVAL_MODE, evals_per_gen, EVAL_FIT_EST_MODE) %>%
summarize(
cov_median = median(pop_optimal_trait_coverage),
cov_mean = mean(pop_optimal_trait_coverage),
n = n()
) %>%
kable()## `summarise()` has grouped output by 'EVAL_MODE', 'evals_per_gen'. You can
## override using the `.groups` argument.| EVAL_MODE | evals_per_gen | EVAL_FIT_EST_MODE | cov_median | cov_mean | n |
|---|---|---|---|---|---|
| cohort | 0.01 | None | 2.0 | 1.9 | 10 |
| cohort | 0.01 | Ancestor | 4.0 | 3.9 | 10 |
| cohort | 0.01 | Relative | 4.0 | 3.9 | 10 |
| cohort | 0.05 | None | 5.0 | 5.0 | 10 |
| cohort | 0.05 | Ancestor | 15.0 | 14.8 | 10 |
| cohort | 0.05 | Relative | 14.0 | 13.7 | 10 |
| cohort | 0.1 | None | 5.5 | 5.5 | 10 |
| cohort | 0.1 | Ancestor | 22.0 | 22.4 | 10 |
| cohort | 0.1 | Relative | 22.0 | 21.6 | 10 |
| cohort | 0.5 | None | 16.0 | 15.9 | 10 |
| cohort | 0.5 | Ancestor | 34.5 | 34.5 | 10 |
| cohort | 0.5 | Relative | 34.0 | 34.2 | 10 |
| down-sample | 0.01 | None | 1.0 | 1.0 | 10 |
| down-sample | 0.01 | Ancestor | 1.5 | 2.5 | 10 |
| down-sample | 0.01 | Relative | 3.5 | 4.7 | 10 |
| down-sample | 0.05 | None | 1.0 | 1.0 | 10 |
| down-sample | 0.05 | Ancestor | 11.5 | 11.0 | 10 |
| down-sample | 0.05 | Relative | 10.0 | 10.0 | 10 |
| down-sample | 0.1 | None | 1.0 | 1.0 | 10 |
| down-sample | 0.1 | Ancestor | 18.0 | 18.1 | 10 |
| down-sample | 0.1 | Relative | 18.0 | 17.1 | 10 |
| down-sample | 0.5 | None | 1.0 | 1.0 | 10 |
| down-sample | 0.5 | Ancestor | 37.5 | 37.6 | 10 |
| down-sample | 0.5 | Relative | 37.5 | 37.5 | 10 |
Next, we perform a Kruskal-Wallis test to determine which comparisons contain statistically significant differences among treatments.
con_obj_kw_test <- con_obj_summary_data %>%
filter(EVAL_MODE != "full") %>%
group_by(EVAL_MODE, evals_per_gen) %>%
kruskal_test(pop_optimal_trait_coverage ~ EVAL_FIT_EST_MODE) %>%
unite(
"comparison_group",
EVAL_MODE,
evals_per_gen,
sep = "_",
remove = FALSE
)
kable(con_obj_kw_test)| comparison_group | EVAL_MODE | evals_per_gen | .y. | n | statistic | df | p | method |
|---|---|---|---|---|---|---|---|---|
| cohort_0.01 | cohort | 0.01 | pop_optimal_trait_coverage | 30 | 25.55066 | 2 | 2.80e-06 | Kruskal-Wallis |
| cohort_0.05 | cohort | 0.05 | pop_optimal_trait_coverage | 30 | 22.72918 | 2 | 1.16e-05 | Kruskal-Wallis |
| cohort_0.1 | cohort | 0.1 | pop_optimal_trait_coverage | 30 | 21.76615 | 2 | 1.88e-05 | Kruskal-Wallis |
| cohort_0.5 | cohort | 0.5 | pop_optimal_trait_coverage | 30 | 20.05082 | 2 | 4.43e-05 | Kruskal-Wallis |
| down-sample_0.01 | down-sample | 0.01 | pop_optimal_trait_coverage | 30 | 15.17863 | 2 | 5.06e-04 | Kruskal-Wallis |
| down-sample_0.05 | down-sample | 0.05 | pop_optimal_trait_coverage | 30 | 20.38430 | 2 | 3.75e-05 | Kruskal-Wallis |
| down-sample_0.1 | down-sample | 0.1 | pop_optimal_trait_coverage | 30 | 20.29663 | 2 | 3.91e-05 | Kruskal-Wallis |
| down-sample_0.5 | down-sample | 0.5 | pop_optimal_trait_coverage | 30 | 20.31895 | 2 | 3.87e-05 | Kruskal-Wallis |
Finally, we perform a pairwise Wilcoxon rank-sum test (using a Holm-Bonferroni correction for multiple comparisons). Note that only results from signific
sig_kw_groups <- filter(con_obj_kw_test, p < 0.05)$comparison_group
con_obj_stats <- con_obj_summary_data %>%
unite(
"comparison_group",
EVAL_MODE,
evals_per_gen,
sep = "_",
remove = FALSE
) %>%
filter(EVAL_MODE != "full" & comparison_group %in% sig_kw_groups) %>%
group_by(EVAL_MODE, evals_per_gen) %>%
pairwise_wilcox_test(pop_optimal_trait_coverage ~ EVAL_FIT_EST_MODE) %>%
adjust_pvalue(method = "holm") %>%
add_significance("p.adj")
kable(con_obj_stats)| EVAL_MODE | evals_per_gen | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif |
|---|---|---|---|---|---|---|---|---|---|---|
| cohort | 0.01 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 3.58e-05 | 0.0008592 | *** |
| cohort | 0.01 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 3.58e-05 | 0.0008592 | *** |
| cohort | 0.01 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 50.0 | 1.00e+00 | 1.0000000 | ns |
| cohort | 0.05 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 9.66e-05 | 0.0015456 | ** |
| cohort | 0.05 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 1.00e-04 | 0.0015456 | ** |
| cohort | 0.05 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 80.0 | 1.90e-02 | 0.1520000 | ns |
| cohort | 0.1 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 1.25e-04 | 0.0016250 | ** |
| cohort | 0.1 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 1.16e-04 | 0.0016240 | ** |
| cohort | 0.1 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 70.5 | 9.60e-02 | 0.5760000 | ns |
| cohort | 0.5 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 1.49e-04 | 0.0017760 | ** |
| cohort | 0.5 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 1.48e-04 | 0.0017760 | ** |
| cohort | 0.5 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 58.0 | 5.56e-01 | 1.0000000 | ns |
| down-sample | 0.01 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 25.0 | 1.50e-02 | 0.1350000 | ns |
| down-sample | 0.01 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 5.0 | 2.27e-04 | 0.0022700 | ** |
| down-sample | 0.01 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 24.0 | 4.90e-02 | 0.3430000 | ns |
| down-sample | 0.05 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 6.25e-05 | 0.0013442 | ** |
| down-sample | 0.05 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 6.16e-05 | 0.0013442 | ** |
| down-sample | 0.05 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 57.5 | 5.92e-01 | 1.0000000 | ns |
| down-sample | 0.1 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 6.25e-05 | 0.0013442 | ** |
| down-sample | 0.1 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 6.29e-05 | 0.0013442 | ** |
| down-sample | 0.1 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 56.0 | 6.75e-01 | 1.0000000 | ns |
| down-sample | 0.5 | pop_optimal_trait_coverage | None | Ancestor | 10 | 10 | 0.0 | 6.11e-05 | 0.0013442 | ** |
| down-sample | 0.5 | pop_optimal_trait_coverage | None | Relative | 10 | 10 | 0.0 | 6.20e-05 | 0.0013442 | ** |
| down-sample | 0.5 | pop_optimal_trait_coverage | Ancestor | Relative | 10 | 10 | 52.0 | 9.08e-01 | 1.0000000 | ns |
# con_obj_stats %>%
# filter(p.adj <= 0.05) %>%
# arrange(
# desc(p.adj)
# ) %>%
# kable()4.3.2 Population-wide satisfactory trait coverage (over time)
contradictory_obj_pop_cov_ts <- ggplot(
con_obj_ts_data,
aes(
x = ts_step,
y = pop_optimal_trait_coverage,
fill = EVAL_FIT_EST_MODE,
color = EVAL_FIT_EST_MODE
)
) +
stat_summary(
geom = "line",
fun = mean
) +
stat_summary(
geom = "ribbon",
fun.data = "mean_cl_boot",
fun.args = list(conf.int = 0.95),
alpha = 0.2,
linetype = 0
) +
scale_fill_bright() +
scale_color_bright() +
facet_wrap(
EVAL_MODE ~ evals_per_gen,
ncol = 1,
labeller = label_both
) +
theme(
legend.position = "bottom"
)
ggsave(
filename = paste0(plot_directory, "contra-obj-ts.pdf"),
plot = contradictory_obj_pop_cov_ts + labs(title="Contradictory objectives"),
width = 10,
height = 15
)contradictory_obj_pop_cov_ts
4.3.3 Phylogeny estimate source distributions
est_source_data %>%
filter(DIAGNOSTIC == "contradictory-objectives") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_self_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of self lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")
ggsave(
filename=paste0(plot_directory, "contra-obj-self-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "contradictory-objectives") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_ancestor_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of ancestor lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")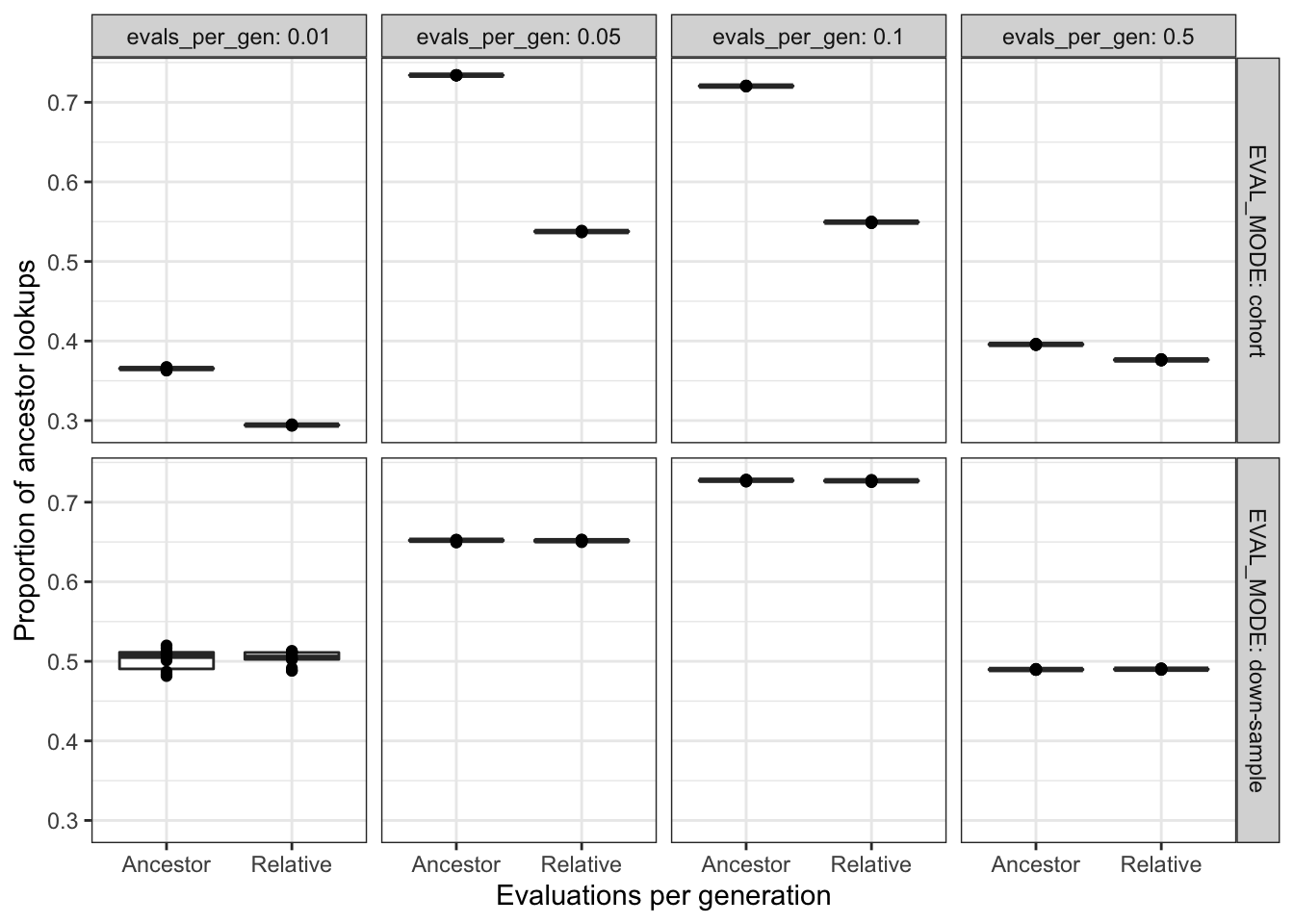
ggsave(
filename=paste0(plot_directory, "contra-obj-ancestor-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "contradictory-objectives") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_descendant_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of descendant lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")
ggsave(
filename=paste0(plot_directory, "contra-obj-descendant-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "contradictory-objectives") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_other_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of other lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")
ggsave(
filename=paste0(plot_directory, "contra-obj-other-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "contradictory-objectives") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_outside_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of outside lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")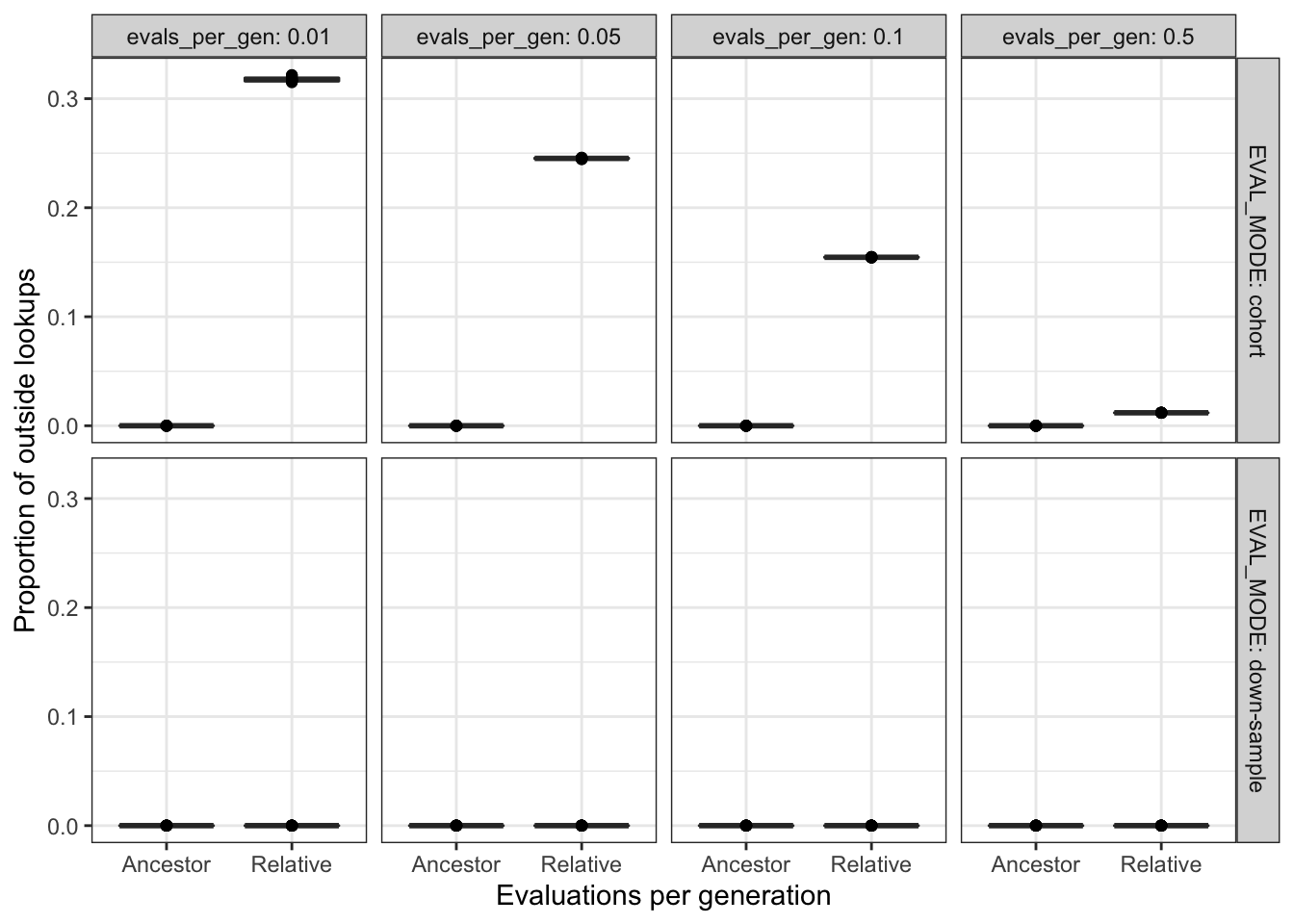
ggsave(
filename=paste0(plot_directory, "contra-obj-outside-lookups.pdf")
)## Saving 7 x 5 in image4.4 Multi-path exploration diagnostic
4.4.1 Maximum aggregate score (final)
explore_final_score_plt <- ggplot(
explore_summary_data,
aes(
x = EVAL_FIT_EST_MODE,
y = max_agg_score,
fill = EVAL_FIT_EST_MODE
)
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
adjust=1.5
) +
geom_point(
mapping=aes(color=EVAL_FIT_EST_MODE),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
# limits = c(-0.5, 100)
) +
scale_fill_bright() +
scale_color_bright() +
facet_grid(
eval_mode_row~evals_per_gen,
# nrow=2,
labeller=label_both
) +
theme(
legend.position = "none",
axis.text.x = element_text(
angle = 30,
hjust = 1
),
panel.border = element_rect(color="gray", size=2)
)
ggsave(
filename = paste0(plot_directory, "explore-final.pdf"),
plot = explore_final_score_plt + labs(title="Multi-path exploration"),
width = 15,
height = 10
)4.4.1.1 Statistical analysis
explore_summary_data %>%
filter(EVAL_MODE != "full") %>%
group_by(EVAL_MODE, evals_per_gen, EVAL_FIT_EST_MODE) %>%
summarize(
score_median = median(max_agg_score),
score_mean = mean(max_agg_score),
n = n()
) %>%
kable()## `summarise()` has grouped output by 'EVAL_MODE', 'evals_per_gen'. You can
## override using the `.groups` argument.| EVAL_MODE | evals_per_gen | EVAL_FIT_EST_MODE | score_median | score_mean | n |
|---|---|---|---|---|---|
| cohort | 0.01 | None | 1971.7450 | 1900.3130 | 10 |
| cohort | 0.01 | Ancestor | 2316.5800 | 1971.5663 | 10 |
| cohort | 0.01 | Relative | 2182.5700 | 2006.6843 | 10 |
| cohort | 0.05 | None | 2401.0150 | 2373.2040 | 10 |
| cohort | 0.05 | Ancestor | 2858.6950 | 2747.0960 | 10 |
| cohort | 0.05 | Relative | 3471.8500 | 3389.0910 | 10 |
| cohort | 0.1 | None | 3075.5150 | 3076.6120 | 10 |
| cohort | 0.1 | Ancestor | 4508.1150 | 4383.9440 | 10 |
| cohort | 0.1 | Relative | 5144.5350 | 5163.0130 | 10 |
| cohort | 0.5 | None | 8187.5000 | 8198.1150 | 10 |
| cohort | 0.5 | Ancestor | 8591.7150 | 8708.0110 | 10 |
| cohort | 0.5 | Relative | 8684.2050 | 8652.1500 | 10 |
| down-sample | 0.01 | None | 580.4215 | 532.1152 | 10 |
| down-sample | 0.01 | Ancestor | 434.8545 | 430.0114 | 10 |
| down-sample | 0.01 | Relative | 449.3640 | 465.0957 | 10 |
| down-sample | 0.05 | None | 396.0890 | 445.1163 | 10 |
| down-sample | 0.05 | Ancestor | 2007.3700 | 1982.4690 | 10 |
| down-sample | 0.05 | Relative | 1777.9000 | 1762.3250 | 10 |
| down-sample | 0.1 | None | 692.7270 | 690.7322 | 10 |
| down-sample | 0.1 | Ancestor | 2423.2200 | 2451.7950 | 10 |
| down-sample | 0.1 | Relative | 2529.6100 | 2542.1340 | 10 |
| down-sample | 0.5 | None | 1499.9800 | 1658.0837 | 10 |
| down-sample | 0.5 | Ancestor | 6976.5950 | 6972.2630 | 10 |
| down-sample | 0.5 | Relative | 7309.9450 | 7120.4160 | 10 |
explore_kw_test <- explore_summary_data %>%
filter(EVAL_MODE != "full") %>%
group_by(EVAL_MODE, evals_per_gen) %>%
kruskal_test(max_agg_score ~ EVAL_FIT_EST_MODE) %>%
mutate(
sig = (p <= 0.05)
) %>%
unite(
"comparison_group",
EVAL_MODE,
evals_per_gen,
sep = "_",
remove = FALSE
)
kable(explore_kw_test)| comparison_group | EVAL_MODE | evals_per_gen | .y. | n | statistic | df | p | method | sig |
|---|---|---|---|---|---|---|---|---|---|
| cohort_0.01 | cohort | 0.01 | max_agg_score | 30 | 0.7045161 | 2 | 7.03e-01 | Kruskal-Wallis | FALSE |
| cohort_0.05 | cohort | 0.05 | max_agg_score | 30 | 15.5380645 | 2 | 4.23e-04 | Kruskal-Wallis | TRUE |
| cohort_0.1 | cohort | 0.1 | max_agg_score | 30 | 25.5509677 | 2 | 2.80e-06 | Kruskal-Wallis | TRUE |
| cohort_0.5 | cohort | 0.5 | max_agg_score | 30 | 5.0348387 | 2 | 8.07e-02 | Kruskal-Wallis | FALSE |
| down-sample_0.01 | down-sample | 0.01 | max_agg_score | 30 | 2.6090323 | 2 | 2.71e-01 | Kruskal-Wallis | FALSE |
| down-sample_0.05 | down-sample | 0.05 | max_agg_score | 30 | 22.3380645 | 2 | 1.41e-05 | Kruskal-Wallis | TRUE |
| down-sample_0.1 | down-sample | 0.1 | max_agg_score | 30 | 19.3780645 | 2 | 6.20e-05 | Kruskal-Wallis | TRUE |
| down-sample_0.5 | down-sample | 0.5 | max_agg_score | 30 | 19.4047630 | 2 | 6.11e-05 | Kruskal-Wallis | TRUE |
expl_sig_kw_groups <- filter(explore_kw_test, p < 0.05)$comparison_group
explore_stats <- explore_summary_data %>%
unite(
"comparison_group",
EVAL_MODE,
evals_per_gen,
sep = "_",
remove = FALSE
) %>%
filter(EVAL_MODE != "full" & comparison_group %in% expl_sig_kw_groups) %>%
group_by(EVAL_MODE, evals_per_gen) %>%
pairwise_wilcox_test(max_agg_score ~ EVAL_FIT_EST_MODE) %>%
adjust_pvalue(method = "holm") %>%
add_significance("p.adj")
kable(explore_stats)| EVAL_MODE | evals_per_gen | .y. | group1 | group2 | n1 | n2 | statistic | p | p.adj | p.adj.signif |
|---|---|---|---|---|---|---|---|---|---|---|
| cohort | 0.05 | max_agg_score | None | Ancestor | 10 | 10 | 27 | 8.90e-02 | 0.2670000 | ns |
| cohort | 0.05 | max_agg_score | None | Relative | 10 | 10 | 4 | 1.30e-04 | 0.0010400 | ** |
| cohort | 0.05 | max_agg_score | Ancestor | Relative | 10 | 10 | 12 | 3.00e-03 | 0.0150000 | * |
| cohort | 0.1 | max_agg_score | None | Ancestor | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| cohort | 0.1 | max_agg_score | None | Relative | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| cohort | 0.1 | max_agg_score | Ancestor | Relative | 10 | 10 | 1 | 2.17e-05 | 0.0001953 | *** |
| down-sample | 0.05 | max_agg_score | None | Ancestor | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| down-sample | 0.05 | max_agg_score | None | Relative | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| down-sample | 0.05 | max_agg_score | Ancestor | Relative | 10 | 10 | 84 | 9.00e-03 | 0.0360000 | * |
| down-sample | 0.1 | max_agg_score | None | Ancestor | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| down-sample | 0.1 | max_agg_score | None | Relative | 10 | 10 | 0 | 1.08e-05 | 0.0001620 | *** |
| down-sample | 0.1 | max_agg_score | Ancestor | Relative | 10 | 10 | 47 | 8.53e-01 | 1.0000000 | ns |
| down-sample | 0.5 | max_agg_score | None | Ancestor | 10 | 10 | 0 | 1.81e-04 | 0.0012670 | ** |
| down-sample | 0.5 | max_agg_score | None | Relative | 10 | 10 | 0 | 1.81e-04 | 0.0012670 | ** |
| down-sample | 0.5 | max_agg_score | Ancestor | Relative | 10 | 10 | 46 | 7.96e-01 | 1.0000000 | ns |
# explore_stats %>%
# filter(p.adj <= 0.05) %>%
# arrange(
# desc(p.adj)
# ) %>%
# kable()4.4.2 Maximum aggregate score (over time)
explore_score_ts <- ggplot(
explore_ts_data,
aes(
x = ts_step,
y = max_agg_score,
fill = EVAL_FIT_EST_MODE,
color = EVAL_FIT_EST_MODE
)
) +
stat_summary(
geom = "line",
fun = mean
) +
stat_summary(
geom = "ribbon",
fun.data = "mean_cl_boot",
fun.args = list(conf.int = 0.95),
alpha = 0.2,
linetype = 0
) +
scale_fill_bright() +
scale_color_bright() +
facet_wrap(
EVAL_MODE ~ evals_per_gen,
ncol = 1,
labeller = label_both
) +
theme(
legend.position = "bottom"
)
ggsave(
filename = paste0(plot_directory, "explore-ts.pdf"),
plot = explore_score_ts + labs(title="Multi-path exploration"),
width = 10,
height = 15
)explore_score_ts
4.4.3 Phylogeny estimate source distributions
est_source_data %>%
filter(DIAGNOSTIC == "multipath-exploration") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_self_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of self lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")
ggsave(
filename=paste0(plot_directory, "explore-self-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "multipath-exploration") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_ancestor_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of ancestor lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")
ggsave(
filename=paste0(plot_directory, "explore-ancestor-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "multipath-exploration") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_descendant_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of descendant lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")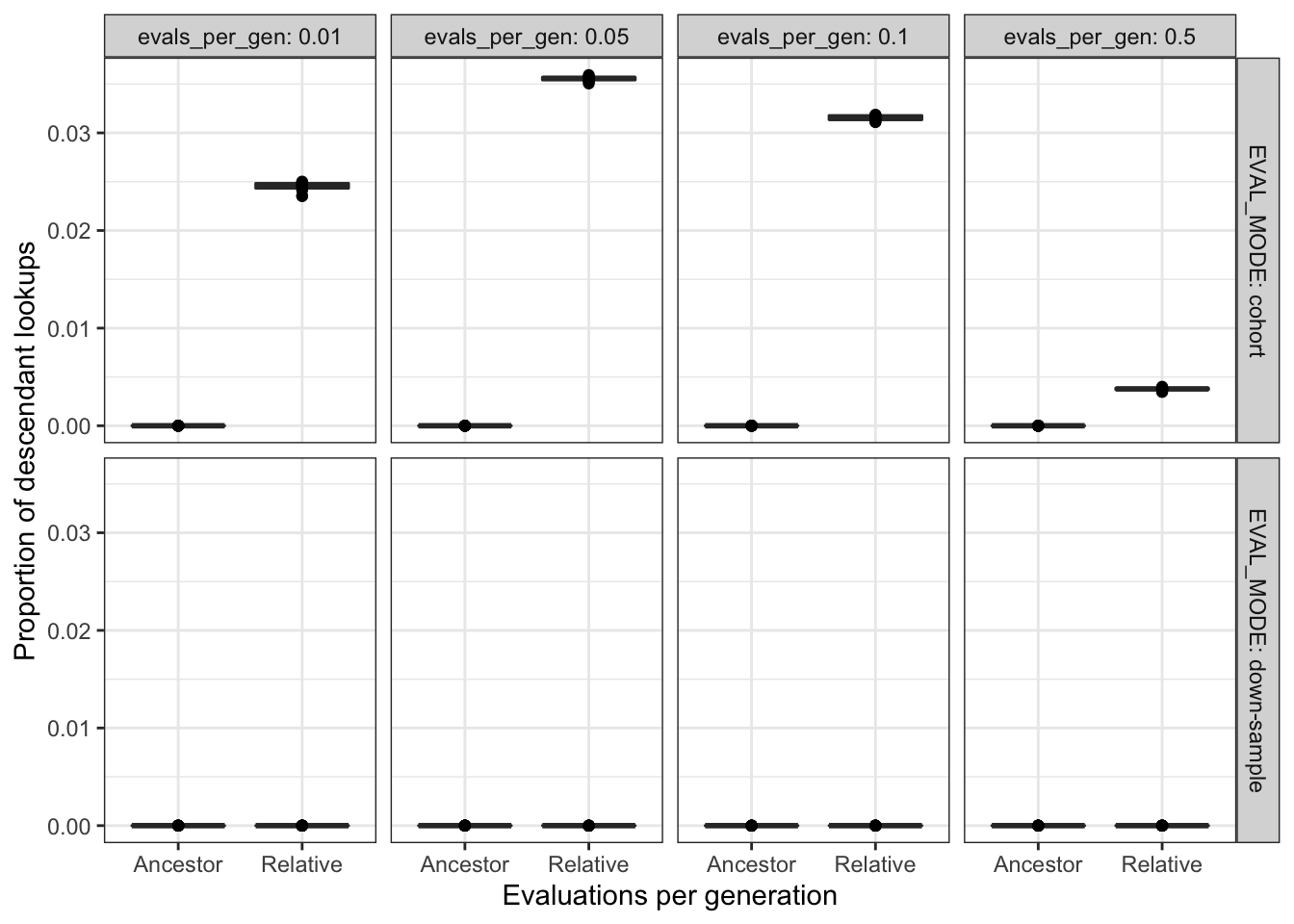
ggsave(
filename=paste0(plot_directory, "explore-descendant-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "multipath-exploration") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_other_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of other lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")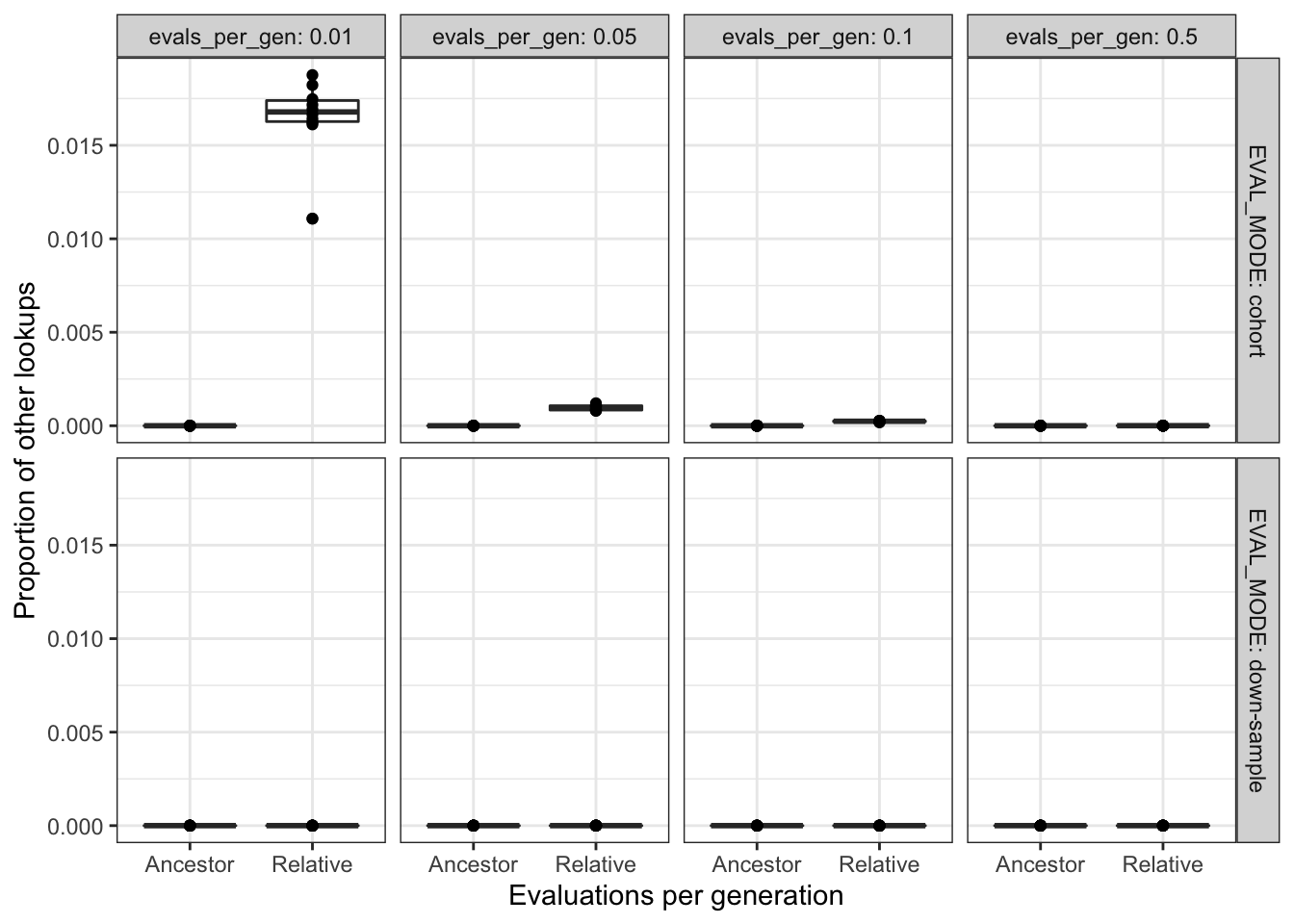
ggsave(
filename=paste0(plot_directory, "explore-other-lookups.pdf")
)## Saving 7 x 5 in imageest_source_data %>%
filter(DIAGNOSTIC == "multipath-exploration") %>%
filter(EVAL_MODE != "full" & EVAL_FIT_EST_MODE != "None") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = prop_outside_lookups
)
) +
geom_boxplot() +
geom_point() +
facet_grid(
cols = vars(evals_per_gen),
rows = vars(EVAL_MODE),
labeller = label_both
) +
scale_y_continuous("Proportion of outside lookups") +
scale_x_discrete("Evaluations per generation") +
theme_bw() +
theme(legend.position = "none")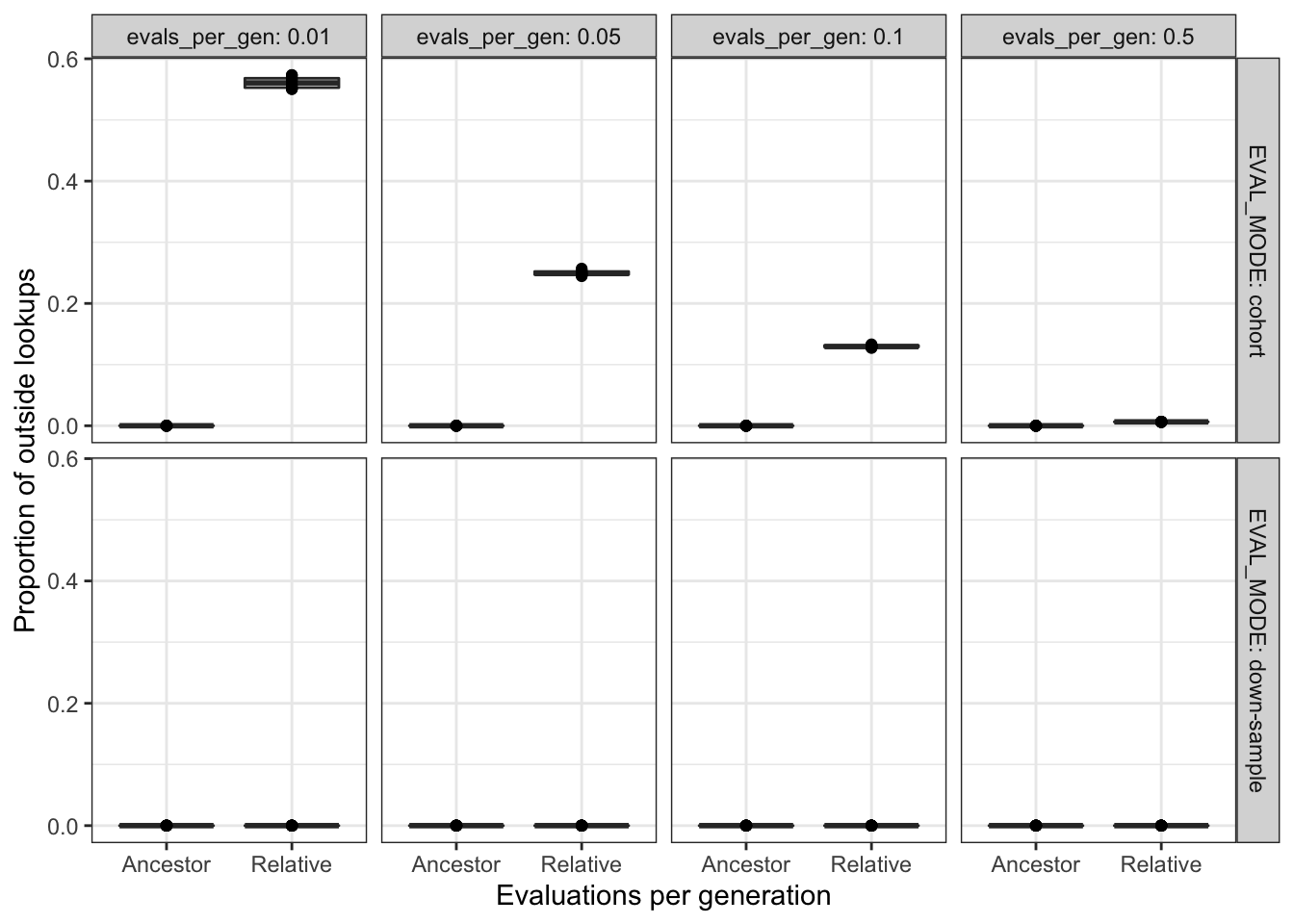
ggsave(
filename=paste0(plot_directory, "explore-outside-lookups.pdf")
)## Saving 7 x 5 in image4.5 Manuscript figures
full_median_size = 1.5
subsample_labeller <- function(subsample_level) {
return(paste("Subsample level:", subsample_level))
}4.5.1 Contradictory objectives
Build plot panels (1 cohort, 1 down-sample)
build_con_obj_plot <- function(eval_mode) {
full_median <- median(
filter(
con_obj_summary_data,
eval_mode_row == eval_mode & EVAL_MODE == "full"
)$pop_optimal_trait_coverage
)
p <- con_obj_summary_data %>%
filter(eval_mode_row == eval_mode & EVAL_MODE != "full") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = pop_optimal_trait_coverage,
fill = EVAL_FIT_EST_MODE
)
) +
geom_hline(
yintercept = full_median,
size = full_median_size,
alpha = 0.7,
color = "black"
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
adjust=1.5
) +
geom_point(
mapping=aes(color=EVAL_FIT_EST_MODE),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
limits = c(-0.5, 50)
) +
scale_fill_bright() +
scale_color_bright() +
facet_wrap(
~ evals_per_gen,
nrow = 1,
labeller = as_labeller(
subsample_labeller
)
) +
labs(
x = "Estimation mode",
y = "Satisfactory trait coverage"
) +
theme(
legend.position = "none",
axis.text.x = element_text(
angle = 30,
hjust = 1
),
panel.border = element_rect(color="gray", size=2)
)
return(p)
}
con_obj_ds_plot <- build_con_obj_plot("down-sample")
con_obj_cohort_plot <- build_con_obj_plot("cohort")Combine panels into single plot.
# Joint title: https://wilkelab.org/cowplot/articles/plot_grid.html
con_obj_title <- ggdraw() +
draw_label(
"Contradictory objectives diagnostic",
fontface = 'bold',
x = 0,
hjust = 0
) +
theme(
# add margin on the left of the drawing canvas,
# so title is aligned with left edge of first plot
plot.margin = margin(0, 0, 0, 7)
)
con_obj_grid <- plot_grid(
con_obj_title,
con_obj_ds_plot +
labs(
title = "Down-sampled lexicase"
) +
theme(axis.title.x = element_blank()),
con_obj_cohort_plot +
labs(
title = "Cohort lexicase"
),
nrow = 3,
ncol = 1,
# align = "h",
labels = c("", "a", "b"),
rel_heights = c(0.075, 1, 1)
)
con_obj_grid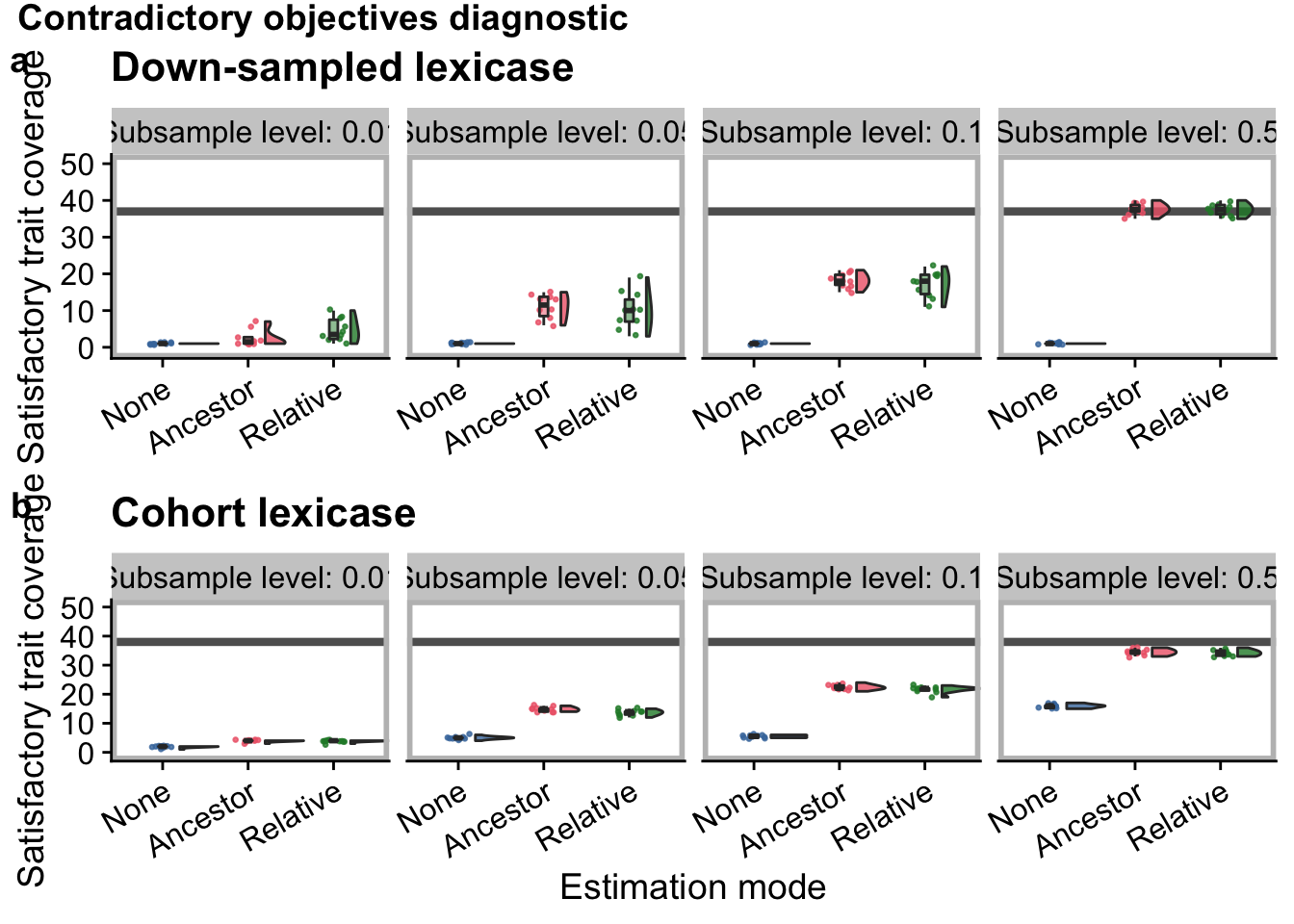
save_plot(
filename = paste0(plot_directory, "2023-05-10-diagnostics-con-obj-final-fig.pdf"),
plot = con_obj_grid,
base_width = 10,
base_height = 8,
dpi = 600
)4.5.2 Multi-path exploration
build_explore_plot <- function(eval_mode) {
full_median <- median(
filter(
explore_summary_data,
eval_mode_row == eval_mode & EVAL_MODE == "full"
)$max_agg_score
)
p <- explore_summary_data %>%
filter(eval_mode_row == eval_mode & EVAL_MODE != "full") %>%
ggplot(
aes(
x = EVAL_FIT_EST_MODE,
y = max_agg_score,
fill = EVAL_FIT_EST_MODE
)
) +
geom_hline(
yintercept = full_median,
size = full_median_size,
alpha = 0.7,
color = "black"
) +
geom_flat_violin(
position = position_nudge(x = .2, y = 0),
alpha = .8,
adjust=1.5
) +
geom_point(
mapping=aes(color=EVAL_FIT_EST_MODE),
position = position_jitter(width = .15),
size = .5,
alpha = 0.8
) +
geom_boxplot(
width = .1,
outlier.shape = NA,
alpha = 0.5
) +
scale_y_continuous(
limits = c(-0.5, 10005)
) +
scale_fill_bright() +
scale_color_bright() +
facet_wrap(
~ evals_per_gen,
nrow = 1,
labeller = as_labeller(
subsample_labeller
)
) +
labs(
x = "Estimation mode",
y = "Max aggregate score"
) +
theme(
legend.position = "none",
axis.text.x = element_text(
angle = 30,
hjust = 1
),
panel.border = element_rect(color="gray", size=2)
)
return(p)
}
explore_ds_plot <- build_explore_plot("down-sample")
explore_cohort_plot <- build_explore_plot("cohort")
explore_ds_plot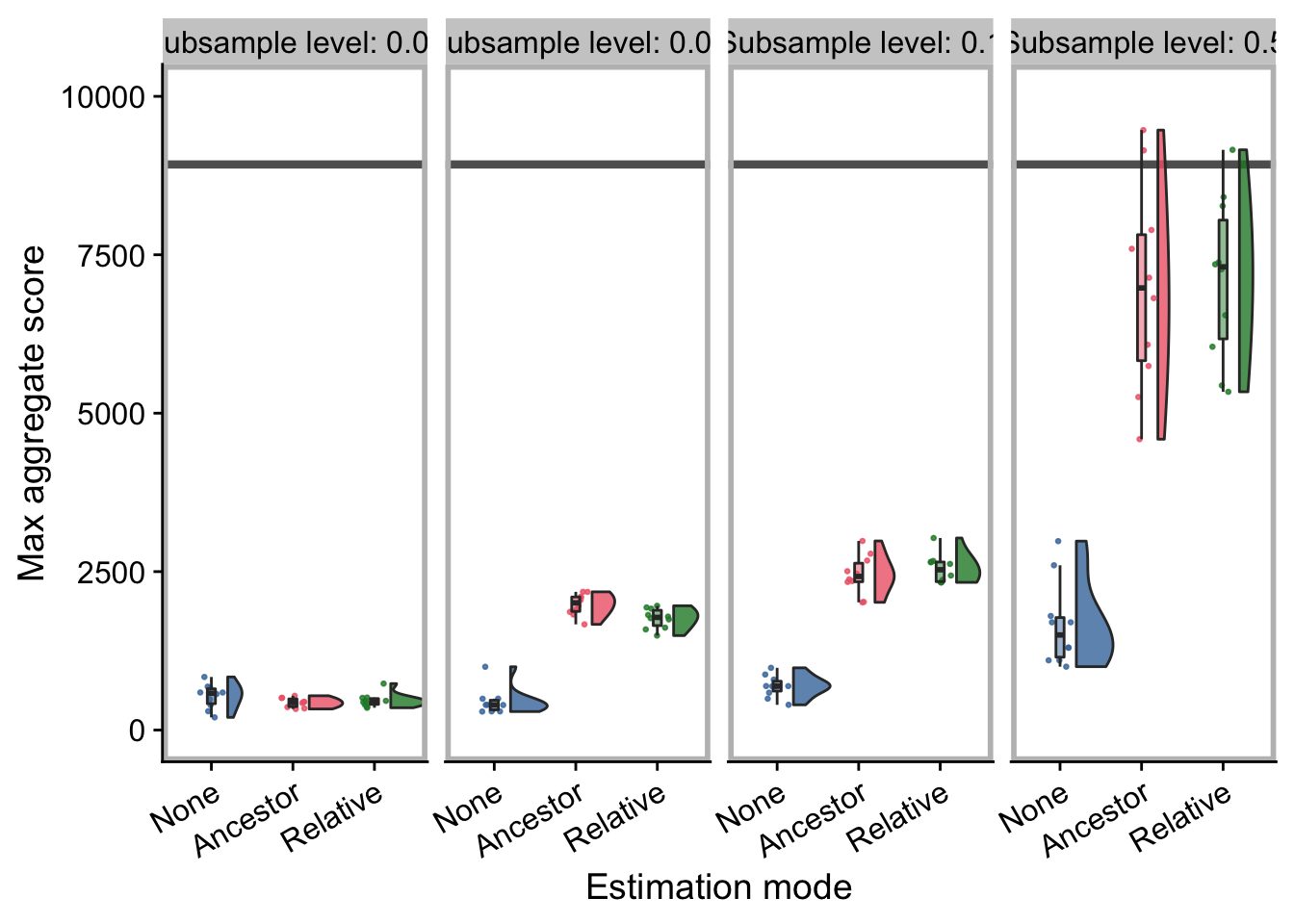
explore_cohort_plot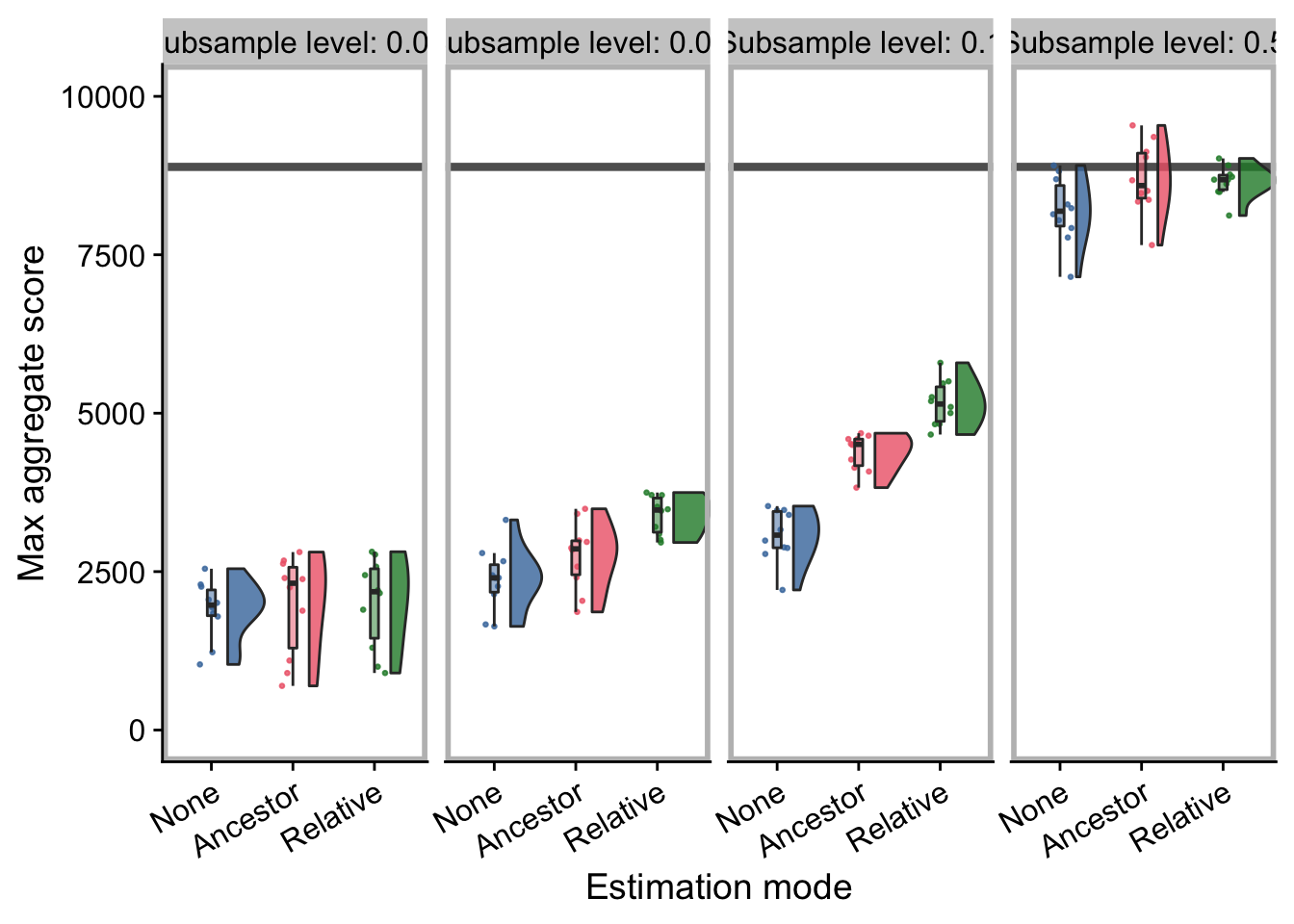
Combine panels into single plot.
# Joint title: https://wilkelab.org/cowplot/articles/plot_grid.html
explore_title <- ggdraw() +
draw_label(
"Multi-path exploration diagnostic",
fontface = 'bold',
x = 0,
hjust = 0
) +
theme(
# add margin on the left of the drawing canvas,
# so title is aligned with left edge of first plot
plot.margin = margin(0, 0, 0, 7)
)
explore_grid <- plot_grid(
explore_title,
explore_ds_plot +
labs(
title = "Down-sampled lexicase"
) +
theme(axis.title.x = element_blank()),
explore_cohort_plot +
labs(
title = "Cohort lexicase"
),
nrow = 3,
ncol = 1,
# align = "h",
labels = c("", "a", "b"),
rel_heights = c(0.075, 1, 1)
)
explore_grid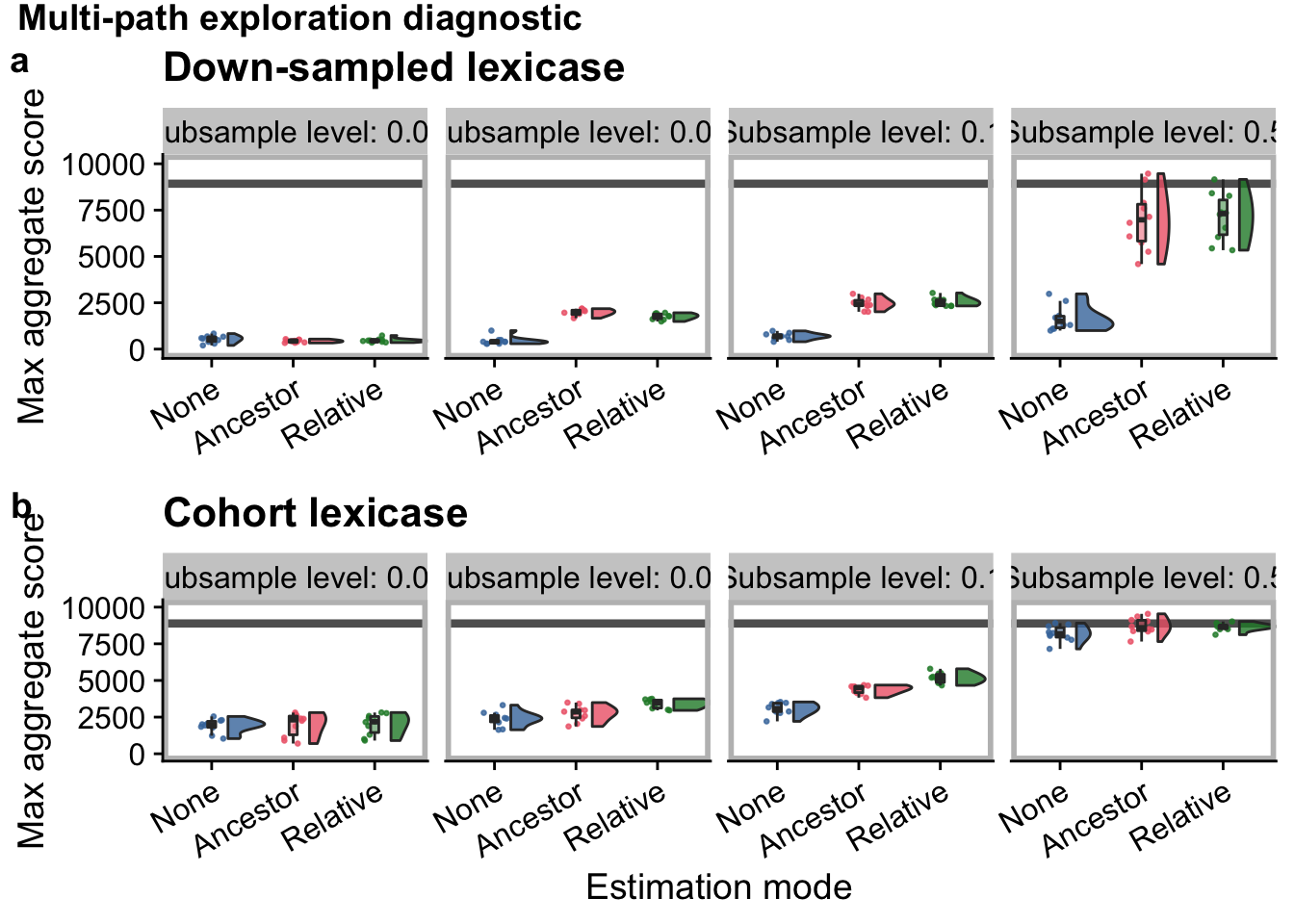
save_plot(
filename = paste0(plot_directory, "2023-05-10-diagnostics-explore-final-fig.pdf"),
plot = explore_grid,
base_width = 10,
base_height = 8,
dpi = 600
)